PhyML, a Montpellier-based software program for tracking the COVID-19 pandemic
The bioinformatics software PhyML, which has been under development for about fifteen years at the Montpellier Laboratory of Computer Science, Robotics, and Microelectronics (Lirmm) and made available to the international scientific community, is now an important tool for tracing the origins of the COVID-19 pandemic and monitoring the virus’s evolution.
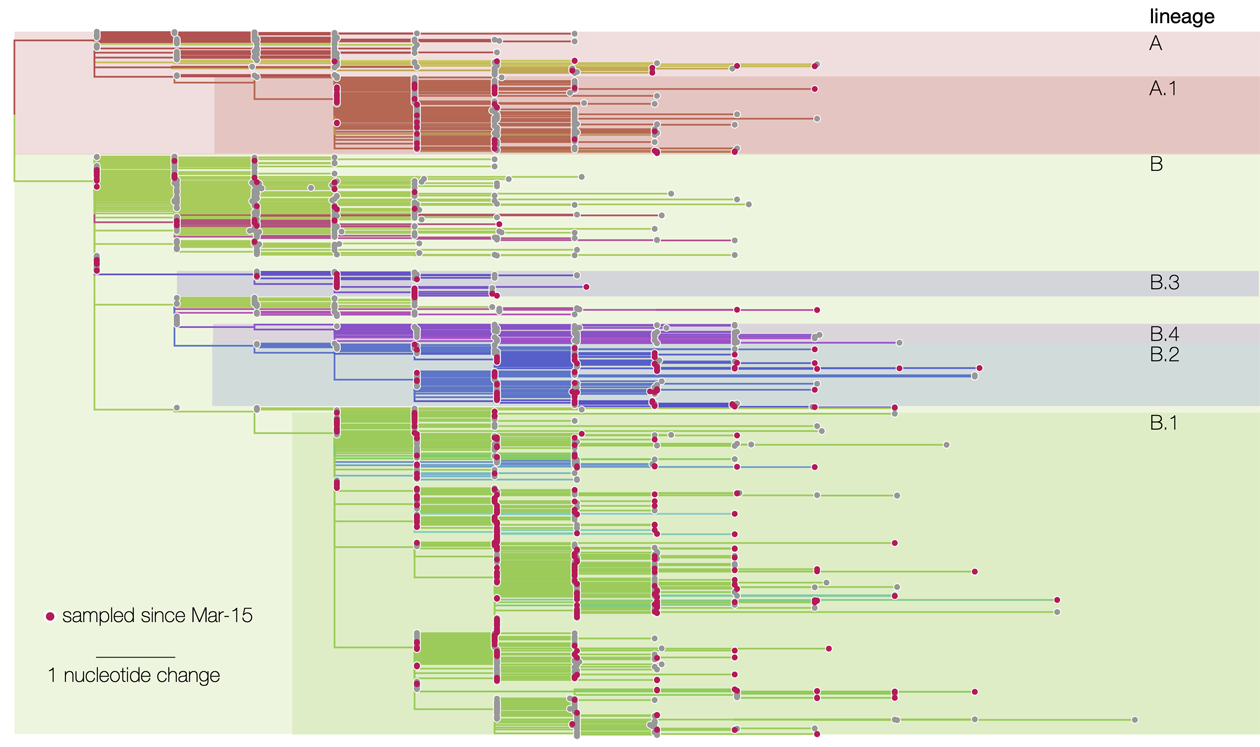
Recent Phylogeny of the SARS-CoV-2 Virus

Stéphane Guindon, CNRS researcher at Lirmm
With the emergence of SARS-CoV-2, the virus responsible for COVID-19, the PhyML software—which was already being used to track more traditional viral outbreaks such as seasonal flu—is now running at full capacity. “We’reworking hard; the software is constantly evolving. We’re implementing algorithms to speed up processing and handle an ever-increasing number of datasets, as well as developing new features…, ”explains Stéphane Guindon, a CNRS researcher at the Montpellier Laboratory of Computer Science, Robotics, and Microelectronics (Lirmm).
Developed in 2003 as part of his doctoral thesis by this bioinformatician and his thesis advisor, Olivier Gascuel, PhyML was among the first free software programs available via the bioinformatics platform hosted by Lirmm. “Wemake our servers available to research laboratories because these phylogenetic analyses are very computationally intensive. Some can take hours, days, or even weeks at times.” Each year, the ATGC platform thus performs approximately 350,000 hours of computation for laboratories located in France, but also—and above all—throughout Europe, China, the United States, and beyond…
Tracing a transmission chain
But what is PhyML used for, and why is it so useful during a pandemic? Because this bioinformatics software is capable of tracing the genealogy—or phylogeny—of any group of organisms with DNA. “Aphylogenetic tree is somewhat like a family tree, which allows us to trace the kinship ties among members of the same family. This ‘genealogy’ is reconstructed here by comparing the genomes of different species. In the case of a virus, we analyze the approximately 30,000 nucleotides that make up its genome in order to trace a chain of transmission.”
The software compared SARS-CoV-2 DNA sequences with all those contained in existing databases, revealing a strong genetic link to a virus found in bats and pangolins. “This isa virus that circulates among bats but does not cause mortality in that species, unlike what we are currently observing in humans, ”notes Stéphane Guindon. Analysis of these sequences also revealed that the current epidemic originated from a single zoonotic event—in other words, a single instance of animal-to-human transmission. “If there had been multiple events, the virus tree would not have the same shape; it would have several subtrees, each corresponding to a transmission of a viral strain from animals to humans.”
Monitoring and Predicting the Spread of the Virus
Phylogenetics has helped answer other important questions about the epidemic. “As early as the beginning of March, the first phylogenetic analyses indicated that the virus’s population size was doubling every 5 to 7 days,” the researcher explains. Phylogenetic analyses using PhyML could also help better predict the long-term evolution of SARS-CoV-2. For example, every year, phylogenetics helps determine which strains of the influenza virus are most likely to circulate the following winter, thereby contributing to the development of effective vaccines.